

La Sindrome di Lennox-Gastaut è un’encefalopatia epilettica dello sviluppo rara e grave ad esordio precoce, caratterizzata dalla triade di deficit cognitivo, tipi multipli di crisi e anomalie tipiche dell’elettroencefalografia (EEG). (Orphanet)
La sindrome di Lennox-Gastaut (LGS) è un disturbo dello sviluppo cerebrale che si accompagna a epilessia e che inizia precocemente sin dai primi anni di vita. Più propriamente è un’Encefalopatia Epilettica, cioè una malattia del cervello dovuta all’epilessia. Questo vuol dire che è l’epilessia stessa ad alterare e a disturbare il normale sviluppo del cervello del bambino.
Per questo la LGS viene anche definita Encefalopatia dello sviluppo ed epilessia (DEE – Developmental Encephalopathy and Epilepsy).
L’ Encefalopatia epilettica è una malattia che danneggia il cervello; le crisi epilettiche che non rispondono al trattamento farmacologico interferiscono con lo sviluppo neuro cognitivo del bambino.
NB: nella EE le ‘crisi’ contribuiscono al deficit cognitivo comportamentale.
L’Epilessia è una malattia neurologica caratterizzata dalla presenza di crisi (manifestazioni motorie, involontarie), che si possono ripetere nel tempo.
NB: Un’unica crisi non fa epilessia!
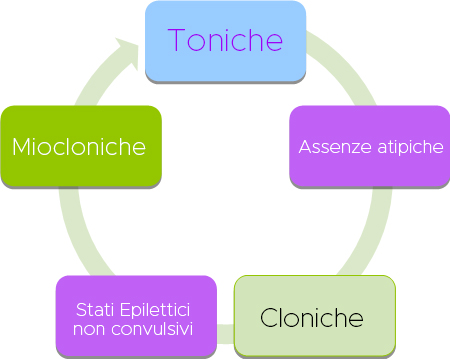
La malattia inizia principalmente tra i 3 ei 5 anni di età e l’intera triade si sviluppa nel tempo. In più della metà dei pazienti, la compromissione cognitiva è presente all’esordio della malattia e peggiora nel tempo. I problemi comportamentali sono comunemente osservati e possono complicare il trattamento.
I tipi di crisi caratteristici includono assenze atipiche e crisi toniche durante il sonno, ma comunemente si verificano crisi atoniche durante la veglia, crisi miocloniche, tonico-cloniche e focali, nonché stato epilettico non convulsivo. Le crisi toniche, miocloniche o atoniche possono portare a cadute improvvise (attacchi di caduta).
NB: Le crisi toniche in sonno e le crisi con caduta (toniche o atoniche), sono caratteristiche della sindrome e ne consentono la diagnosi.
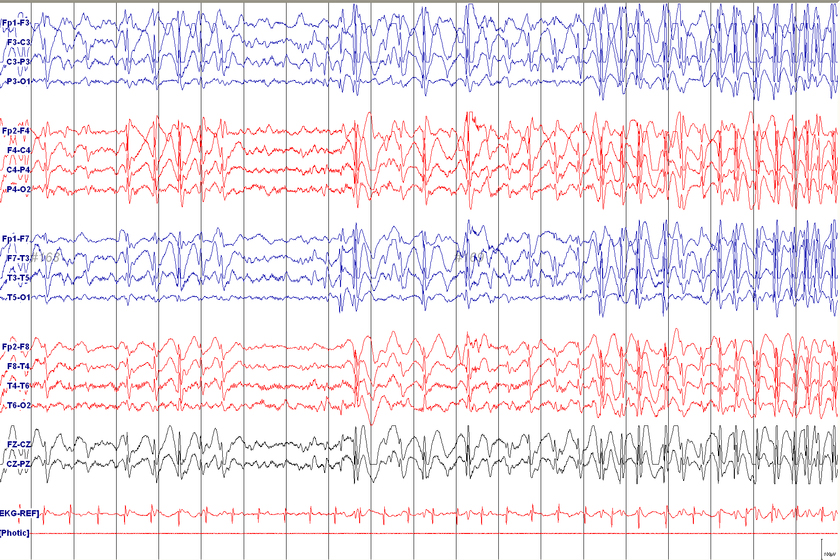
Tramite una valutazione clinica approfondita, un’anamnesi dettagliata del paziente e attraverso tecniche di imaging avanzate, come l’Elettroencefalogramma (EEG) e la Risonanza Magnetica (RMN).
In particolare i segni per diagnosticare la LGS sono:
Anamnesi dettagliata sin dai primi giorni di vita del paziente, alla ricerca di qualsiasi possibile anomalia.
Test medico per misurare l’attività elettrica del cervello.
Nella maggior parte degli EEG con LGS si osservano due elementi tipici:
La risonanza magnetica serve a trovare eventuali cause strutturali di LGS e può rilevare:
Non esiste un pattern RMN caratteristico per LGS. La risonanza magnetica potrebbe anche risultare normale.
I test genetici per la LGS attualmente sono tre:
Il trattamento è difficile, la LGS è farmacoresistente e più dell’80% dei pazienti continua a presentare crisi nonostante le terapie.
Le possibilità terapeutiche sono:
I farmaci più utilizzati sono:
Viene rivalutata l’efficacia del farmaco somministrato dopo 2-3 mesi
Terapia Chirurgica
(VNS stimolazione del nervo vago, Callosotomia totale o parziale), (DBS stimolazione cerebrale profonda)
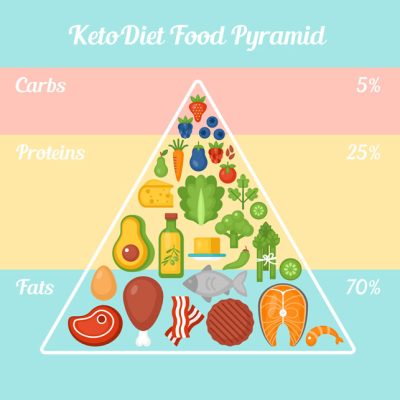
La dieta chetogenica
Un particolare regime alimentare, ricco di grassi e privo di carboidrati e proteine, condotto sotto stretto controllo medico, che ha come obiettivo quello di indurre e mantenere uno stato chetonico
Impostazioni privacy
Questo sito utilizza i cookie per migliorare la tua esperienza di navigazione su questo sito.
Visualizza la Cookie Policy Visualizza l'Informativa Privacy
YouTube è un servizio di visualizzazione di contenuti video gestito da Google Ireland Limited e permette a questo Sito Web di integrare tali contenuti all’interno delle proprie pagine.
Questo widget è impostato in modo che YouTube non salvi informazioni e cookie inerenti agli Utenti su questo Sito Web, a meno che non riproducano il video.
Luogo del trattamento: Irlanda - Privacy Policy
Cloudflare Turnstile è un servizio di protezione dallo SPAM fornito da CloudFlare Inc.
Luogo del trattamento: Stati Uniti - Privacy Policy
Google Fonts è un servizio per visualizzare gli stili dei caratteri di scrittura gestito da Google Ireland Limited e serve ad integrare tali contenuti all’interno delle proprie pagine.
Luogo del trattamento: Irlanda - Privacy Policy
Gravatar è un servizio di visualizzazione di immagini gestito da Automattic Inc. che permette a Automattic Inc. di integrare tali contenuti all’interno delle proprie pagine.
Luogo del trattamento: Stati Uniti - Privacy Policy
